16, 17-二羟基紫蒽酮 生产工艺 128-59-6
CAS Name: Dinaphtho[1,2,3-cd:3’,2’,1’-lm]perylene-5,10-dione, 16,17-dihydroxy-
Anthra[9,1,2-cde]benzo[rst]pentaphene-5,10-dione, 16,17-dihydroxy- or Violanthron, 16,17-dihydroxy-
用途: 还原蓝 16 (C.I. 71200). 还原蓝 53 (C.I. 71205). 还原绿 1 (C.I. 59825) = 颜料绿 47.
还原绿 2 (C.I. 59830) = 颜料绿 54. 溶剂绿 6 (C.I. 59845).
合成路线.
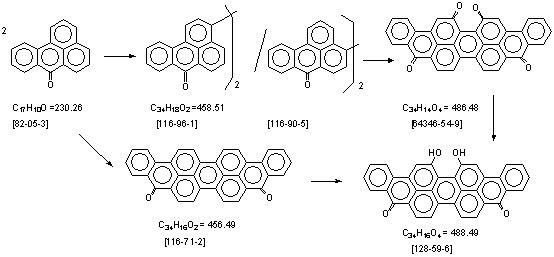
生产工艺汇编: (译自德文PB.报告)
1. 3,3’-联苯绕蒽酮. [116-96-1]. 参考 DE. 589079. 1937年8月2日. 译自PB. 25628. 3740-1.
1). 粗品合成.
300 g 96%硫酸中, 于100C加入50 g升华苯绕蒽酮, 搅半小时至完全溶解, 冷至00C, 加入软锰矿粉硫酸悬浮液. 它由45 g 90-92% 经磨细的软锰矿粉, 在450 g 96%硫酸中, 于室温搅拌12小时配成.
加毕, 在0 –100C搅拌5小时, 最后将料液放入到加有90% 350波美亚硫酸氢钠液的7500 g水中, 煮沸5分钟, 过滤, 用热水洗至中性.
滤饼于加有20 g 400波美液碱的1000 g水中煮沸20分钟, 趁热过滤, 用热水洗至中性, 滤液为无色, 烘干, 得粗品 49 –50 g.
2). 精制.
100 g粗品, 加入600 g邻二氯苯, 加热煮沸一小时, 冷至500C, 过滤, 滤饼用耙式干燥器蒸出邻二氯苯, 并烘干 (实验室可用乙醇洗除邻二氯苯). 得量. 40 –45 g.
备注: (1). 软锰矿粉悬浮液, 可以不冷却, 因苯绕蒽酮已足够冷.
(2). 软锰矿粉量, 可在35 –55 g之间.
(3). 配制后的软锰矿悬浮液, 可放置8天, 不影响使用.
(4). 反应温度过高, 20 –300C, 对反应不利.
2. 3,3’-联苯绕蒽酮. [116-96-1]. 1938年9月20日. 译自PB. 25628. 3739.
450 g 96%硫酸中, 加入45 g二氧化锰, 于室温搅拌过夜, 次日加入500 g 96%硫酸, 冷至00C, 在0 –100C, 在约一小时内, 加入50 g升华苯绕蒽酮, 加毕, 在00C搅拌5小时.
将反应液滴加入加有90 g亚硫酸氢钠的750 ml水中, 煮沸5分钟, 过滤, 热水洗至中性.
将湿滤饼加入到加有125 g 50%液碱的2500 ml水中, 加热到650C, 加入33 g保险粉, 再在650C搅拌15分钟, 过滤, 滤饼用 ( 1000 ml水 + 50 ml液碱 +15 g保险粉) 还原液洗涤, 再用热水洗, 烘干.
得量:46 g.
3. 4,4’-联苯绕蒽酮. [116-90-5]. 生产工艺. 1931年5月28日. 译自PB. 25624. 1031-9 (甲醇 / 碱)
生产设备. 略!
生产工艺 (操作步骤):
在配制锅内, 予先通过计量槽加入200 kg蒸镏过的甲醇, 手工加入200 kg升华粉碎过的苯绕蒽酮, 搅拌一小时, 与此同时, 在3立方米反应锅内加入800 kg苛性钾 (90%), 用料泵, 通过计量槽加入250 kg甲醇 (98 – 100%), 在1100C回流加热到物料均匀为止, 然后在半小时内接受配制好的苯绕蒽酮液, 无甲醇蒸出, 配制锅用少量甲醇 (约15 kg) 冲洗, 合倂后, 加热到110 –1150C, 保温6小时, 当保温到4小时后, 开始(取样I)测终点, 终点到后, 在4 – 5小时内, 由计量槽加入1200 kg热水, 同时蒸出甲醇, 收集在2.9立方米受槽内, 在加水的同时, 需加入30 kg 次氯酸钠液 (12.5%), 蒸镏到减压仍无甲醇蒸出为止, 物料通过冷却锅加到11.5立方米稀释锅内, 锅内予先加有, 约2000 L水, 受料后, 用直接蒸汽加热至沸, 保持一小时左右, 然后用约6000 L水稀释, 于40 –500C压滤, 热水洗到中性, 于耙式干燥器中, 在1300C 干燥 (取样II)
得量: 200 kg升华苯绕蒽酮, 得205 –210 kg联苯绕蒽酮 (78 – 83%) =164 kg, 100%
收率: 82.3%
操作要点:
甲醇要尽可能的纯, 含量不低于95 – 96%, 因此蒸镏出来的甲醇, 必须进行精镏.
对200 kg苯绕蒽酮, 甲醇最大用量, 不要超过500 kg.
所用苯绕蒽酮, 应只含微量蒽醌 (大结晶升华物). 予先配制苯绕蒽酮甲醇液, 可减少物料粘壁, 从而减少苯绕蒽酮的损失.
熔融温度不要超过1200C, 因为在1200C以上会产生紫蒽酮 (即C.I. 还原蓝20).
不正常原因:
甲醇含水量高 (> 5%). 甲醇用量多了, 有利于紫蒽酮的生成, 也就是说, 影响联苯绕蒽酮的生成.
升华苯绕蒽酮中, 蒽醌含量高 (细结晶升华物), 收率也要降低.
生产控制点:
取样I: 反应4小时后, 取样, 于水中析出, 煮沸, 过滤, 洗涤, 干燥, 测熔点, 应不低于312 –50C.
苯绕蒽酮含量测定: 取0.1 g干粉, 于100 ml 96%硫酸中溶解, 取出1 ml, 用硫酸稀释到100 ml, 于汞灯下, 与标准液比较. 测定纯度.
紫蒽酮含量测定: 取干粉, 于稀碱液中, 用保险粉还原, 过滤, 点滤液于滤纸上, 蓝色表明有紫蒽酮.
取样II: 成品干粉, 按上面同样方法, 测熔点,化苯绕蒽酮和紫蒽酮含量. 成品为绿色 (不显深紫色)
原料质量控制:
苯绕蒽酮熔点: 167 –1680C. 其碱性保险粉还原, 应微显黄红色.
测蒽醌含量, 灰份.
成品质量控制:
成品熔点: 315 –3170C. 溶于浓硫酸为微带荧光的深红色 (苯绕蒽酮).
纯度测定方法:
于1立升三口瓶中, 加入300 ml三氯苯,50 g联苯绕蒽酮粗粉, 加热回流煮沸一小时, 然后慢慢冷却, 先用空气冷, 最后用水冷到室温, 放置过夜 (至少放置15小时), 于已称重过的滤杯中过滤, 用300 ml三氯苯洗涤, 再用乙醇洗出三氯苯, (最好用乙醇处理, 而后过滤). 干燥.
纯度约为 82 – 86%, 减去灰份, 实际纯度为79 – 83%.
这一分析法是可行的, 但不完美, 因在三氯苯中, 结晶不完全.
溶剂回收: 蒸镏得到的甲醇-水混合液, 于精镏塔中蒸镏回收.
甲醇损耗率: 20-25%
生产经验:
在温度 100 –1200C的甲醇苛性钾中, 反应3 – 4小时, 即可, 如低于1200C, 反应增加到6 – 10小时也行, 不会有紫蒽酮生成.
合适的反应时间为4 – 6小时, 温度为1150C. 在同样条件下, 使用乙醇苛性钾反应, 产品不纯.
甲醇用量, 只能在小范围内变动, 甲醇用量大于2.5倍 (以苯绕蒽酮计), 低温反应会有紫蒽酮生成, 同时使生成联苯绕蒽酮的速度减慢., 甲醇用量低于规程, 搅拌会困难. 同样, 甲醇含水量 >5%, 也不行, 一方面是生成紫蒽酮, 另一方面是反应不完全.
加次氯酸钠, 目的是使隐色体氧化.
专利: DE. 407838. US. 1564423.






![C.I. 酸性蓝145 (C.I. 62070) 生产工艺 CAS号[6408-80-6]](/chenzhongyuan/Upload/ArticlePicture/IMG_29062016_105104.png "C.I. 酸性蓝145 (C.I. 62070) 生产工艺 CAS号[6408-80-6] 2016/6/29")
![C.I. 酸性橙51 (C.I. 26500) 生产工艺 CAS号 [8003-88-1]](/chenzhongyuan/Upload/ArticlePicture/IMG_21062016_132801.png "C.I. 酸性橙51 (C.I. 26500) 生产工艺 CAS号 [8003-88-1] 2016/6/21")
![CAS号 [109-89-7] 生产工艺。 二乙胺](/chenzhongyuan/Style/Blue/Style_Bullets_Picture.gif "CAS号 [109-89-7] 生产工艺。 二乙胺 2020/7/20")