CAS号 [128-59-6] 生产工艺增补。 16, 17-二羟基紫蒽酮
增补说明:CAS号 [128-59-6],已在2018年4月19日上网。当时主要介绍美国人的译文。今天在整理以前在沈阳院工作时所翻译的,美国人所介绍的德文生产工艺原件,发现还有一点参考价值,至少国内染料行业无人看过!现整理抄录如下:
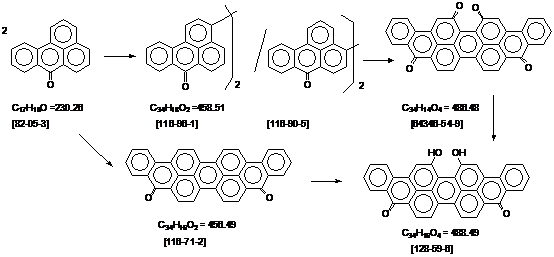
合成路线.
生产工艺汇编: (译自德文PB.报告,译文仅供参考!)
1.
3,3’-联苯绕蒽酮. [116-96-1]. 引用专利 DE. 589079. 1937年8月2日. 译自PB. 25628. 3740-1.
1). 粗品合成.
300
g
96%硫酸中,
于10℃加入50 g升华苯绕蒽酮, 搅半小时至完全溶解, 冷至0℃,
加入软锰矿粉硫酸悬浮液.
它由45 g 90-92% 经磨细的软锰矿粉, 在450
g
96%硫酸中,
于室温搅拌12小时配成.
加毕, 在0 – 10℃搅拌5小时, 最后将料液放入到加有90% 350波美亚硫酸氢钠液的7500
g水中, 煮沸5分钟, 过滤, 用热水洗至中性.
滤饼于加有20
g
400波美液碱的1000 g水中煮沸20分钟, 趁热过滤, 用热水洗至中性, 滤液为无色, 烘干, 得粗品 49 – 50 g.
2). 精制.
100
g粗品, 加入600
g邻二氯苯, 加热煮沸一小时, 冷至50℃,
过滤,
滤饼用耙式干燥器蒸出邻二氯苯,
并烘干
(实验室可用乙醇洗除邻二氯苯). 得量. 40 – 45 g.
备注: (1). 软锰矿粉悬浮液, 可以不冷却, 因苯绕蒽酮已足够冷.
(2). 软锰矿粉量, 可在35 – 55 g之间.
(3). 配制后的软锰矿悬浮液, 可放置8天, 不影响使用.
(4). 反应温度过高, 20 – 30℃, 对反应不利.
2.
3,3’-联苯绕蒽酮. [116-96-1]. 1938年9月20日. 译自PB. 25628. 3739.
450
g
96%硫酸中,
加入45 g二氧化锰, 于室温搅拌过夜, 次日加入500
g
96%硫酸,
冷至0℃,
在0
– 10℃, 在约一小时内, 加入50 g升华苯绕蒽酮, 加毕, 在0℃搅拌5小时.
将反应液滴加入加有90
g亚硫酸氢钠的750 ml水中, 煮沸5分钟, 过滤, 热水洗至中性.
将湿滤饼加入到加有125
g
50%液碱的2500
ml水中,
加热到65℃,
加入33 g保险粉, 再在65℃搅拌15分钟, 过滤, 滤饼用 ( 1000 ml水 + 50 ml液碱 + 15 g保险粉) 还原液洗涤, 再用热水洗, 烘干.
得量: 46 g.
3.
4,4’-联苯绕蒽酮. [116-90-5]. 生产工艺. 1931年5月28日. 译自PB. 25624. 1031-9 (甲醇 / 碱)
生产设备. 略!
生产工艺 (操作步骤):
在配制锅内, 予先通过计量槽加入200
kg蒸镏过的甲醇, 手工加入200
kg升华粉碎过的苯绕蒽酮, 搅拌一小时, 与此同时, 在3立方米反应锅内加入800
kg苛性钾 (90%), 用料泵, 通过计量槽加入250
kg甲醇 (98 – 100%), 在110℃回流加热到物料均匀为止, 然后在半小时内接受配制好的苯绕蒽酮液, 无甲醇蒸出, 配制锅用少量甲醇 (约15
kg)
冲洗,
合倂后,
加热到110
– 115℃, 保温6小时, 当保温到4小时后, 开始(取样I)测终点, 终点到后, 在4 – 5小时内, 由计量槽加入1200
kg热水, 同时蒸出甲醇, 收集在2.9立方米受槽内, 在加水的同时, 需加入30
kg
次氯酸钠液
(12.5%), 蒸镏到减压仍无甲醇蒸出为止, 物料通过冷却锅加到11.5立方米稀释锅内, 锅内予先加有, 约2000
L水, 受料后, 用直接蒸汽加热至沸, 保持一小时左右, 然后用约6000
L水稀释, 于40 – 50℃压滤, 热水洗到中性, 于耙式干燥器中, 在130℃
干燥
(取样II)
得量: 200
kg 升华苯绕蒽酮, 得205 – 210 kg联苯绕蒽酮 (78 – 83%) = 164 kg , 100%
收率: 82.3%
操作要点:
甲醇要尽可能的纯, 含量不低于95 – 96%, 因此蒸镏出来的甲醇, 必须进行精镏.
对200
kg苯绕蒽酮, 甲醇最大用量, 不要超过500
kg.
所用苯绕蒽酮, 应只含微量蒽醌 (大结晶升华物). 予先配制苯绕蒽酮甲醇液, 可减少物料粘壁, 从而减少苯绕蒽酮的损失.
熔融温度不要超过120℃,
因为在120℃以上会产生紫蒽酮 (即C.I. 还原蓝20).
不正常原因:
甲醇含水量高 (> 5%). 甲醇用量多了, 有利于紫蒽酮的生成, 也就是说, 影响联苯绕蒽酮的生成.
升华苯绕蒽酮中, 蒽醌含量高 (细结晶升华物), 收率也要降低.
生产控制点:
取样I: 反应4小时后, 取样, 于水中析出, 煮沸, 过滤, 洗涤, 干燥, 测熔点, 应不低于312 – 5℃.
苯绕蒽酮含量测定: 取0.1
g干粉, 于100 ml 96%硫酸中溶解, 取出1 ml, 用硫酸稀释到100 ml, 于汞灯下, 与标准液比较. 测定纯度.
紫蒽酮含量测定: 取干粉, 于稀碱液中, 用保险粉还原, 过滤, 点滤液于滤纸上, 蓝色表明有紫蒽酮.
取样II: 成品干粉, 按上面同样方法, 测熔点,化苯绕蒽酮和紫蒽酮含量. 成品为绿色 (不显深紫色)
原料质量控制:
苯绕蒽酮熔点: 167 – 168℃.
其碱性保险粉还原,
应微显黄红色.
测蒽醌含量, 灰份.
成品质量控制:
成品熔点: 315 – 317℃.
溶于浓硫酸为微带荧光的深红色
(苯绕蒽酮).
纯度测定方法:
于1立升三口瓶中, 加入300 ml三氯苯, 50 g 联苯绕蒽酮粗粉, 加热回流煮沸一小时, 然后慢慢冷却, 先用空气冷, 最后用水冷到室温, 放置过夜 (至少放置15小时), 于已称重过的滤杯中过滤, 用300 ml三氯苯洗涤, 再用乙醇洗出三氯苯, (最好用乙醇处理, 而后过滤). 干燥.
纯度约为 82 – 86%, 减去灰份, 实际纯度为79 – 83%.
这一分析法是可行的, 但不完美, 因在三氯苯中, 结晶不完全.
溶剂回收: 蒸镏得到的甲醇-水混合液, 于精镏塔中蒸镏回收.
甲醇损耗率: 20-25%
生产经验:
在温度 100 – 120℃的甲醇苛性钾中, 反应3 – 4小时, 即可, 如低于120℃,
反应增加到6
– 10小时也行,
不会有紫蒽酮生成.
合适的反应时间为4 – 6小时, 温度为115℃.
在同样条件下,
使用乙醇苛性钾反应,
产品不纯.
甲醇用量, 只能在小范围内变动, 甲醇用量大于2.5倍 (以苯绕蒽酮计), 低温反应会有紫蒽酮生成, 同时使生成联苯绕蒽酮的速度减慢., 甲醇用量低于规程, 搅拌会困难. 同样, 甲醇含水量 >5%, 也不行, 一方面是生成紫蒽酮, 另一方面是反应不完全.
加次氯酸钠, 目的是使隐色体氧化. 引用专利:DE. 407838; US.
1564423.
4.
联苯绕蒽酮 J [116-90-5] 生产工艺. 1937年6月10日.
译自
PB.
70057. 8393-8405. (异丁醇 / 碱)
理论投得量: 200 (100%)
à 199.2 100 (100%) à
99.6
实际投得量: 200 (100%)
à 164 (100%) 100 (100%) à
82 (100%) 收率: 82.3%
生产周期及日产量: 24小时. 日产量约200
公斤 (82%). 合164公斤 (100%)
生产设备: 略!
生产工艺 (操作步骤):
通过流量计, 向3立方米碱熔锅内打入385公斤 = 480立升纯异丁醇, 由加料器加入500公斤 (88-90%) 粉碎好的苛性钾和 60公斤无水醋酸钠, 此时以10转 / 分速度搅拌, 汽门完全打开, 加热到内温140 – 145℃
(约需2小时), 将蒸镏管线与回流冷凝器相接, 加热开始后可慢慢提高搅拌转速, 关键是马达电流不要大于7安培, 于140 – 145℃搅拌一小时, 此时异丁醇有蒸出, 而不回流.
用冷却水, 将物料冷到90℃
(约需2小时), 大部分苛性钾结皮析出在锅壁上, 用蒸汽加热, 使其重新熔化, 趁锅壁的热, 用长铲将其全部铲除, 内温开始升高, 停止加热, 在100℃,
于一小时内,
通过篩网
(1目)
人工加入200 公斤经升华粉碎好的苯绕蒽酮, 加料时物料已很粘, 加入100公斤苯绕蒽酮后, 电流可达8安培, 不停马达, 用降低搅拌转速的办法使电流维持在7安培, 在继续加苯绕蒽酮的过程中, 搅拌只能维持在15 – 20 转/分, 因反应放热, 加完时, 内温可达112℃,
在此温度下再搅拌一小时
(取样),
不用等取样结果,
往下进行处理,
经流量计在5分钟内向粘稠的反应物内打入100公斤 = 120立升纯异丁醇, 加完, 温度自行降到105 – 110℃,
物料变稀,
全速开动搅拌,
用冷水将物料冷到90℃
(约需2小时), 当冷到95℃时, 物料又开始变稠, 需再降低搅拌转速, 于一小时内, 由计量槽加入1000立升饮用水, 同时夹套通冷水, 物料立即变稀, 再全速搅拌, 加完冷水, 温度降到40℃,
进一步冷却到25
– 30℃ (约需2小时), 再搅拌半小时, 静止, 物料分为二层, 下层为30%左右的苛性钾水溶液, 呈红色, 上层为联苯绕蒽酮和联苯绕蒽酮隐色体在异丁醇中的混合液. 下层不含异丁醇, 分层除去, 为此, 将出口管线与计量槽接通, 开阀, 让1000立升碱液流入计量槽中, 留在锅内的绿色物料层, 在一小时内加入400立升分离水或饮用水, 同时在计量槽中, 将600立升分离水或饮用水与80公斤漂白液 (约12%)配混, 也在一小时内将其加到锅内, 搅拌半小时, 将蒸镏管线与冷凝器, 分离器接通, 加热至沸 (约需一小时), 蒸出异丁醇, 水恒沸混合液 (内温90 – 92℃),
再由计量槽加入与蒸出量相等的分离水 (约需3小时).
将分离液 (蒸出液) 分层, 上层为含有83%的异丁醇层, 放入含水醇的贮槽, 下层为含5%异丁醇的水层, 可再回到锅内.
当大部分异丁醇蒸出后, 温度约到98℃,
有冲料危险,
因此,
必须随时观察锅上蒸镏管线中的视镜,
发现有物料泳上视镜,
应立即搅拌片刻,
消除冲料.
当内温到101
– 102℃, 蒸镏完毕, 在此温度下搅拌约半小时, 不用加分离水, 此时冷凝器下面的视镜应无异丁醇出现, 将经蒸镏后的浅绿色物料 (约1500立升)压到予先加有2000立升热水的22000立升稀释锅内, 用热水洗锅, 最后用热水稀释到8000立升, 用直接蒸汽加热到90 – 95℃
(约需2小时), 在此温度下搅拌一小时, 压料到铁压滤机中过滤, 热水洗涤滤饼, 湿滤饼于耙式干燥器中减压干燥, 出料前夹套通冷水, 干燥器内通氮气.
得量: 200公斤升华苯绕蒽酮, 得200公斤联苯绕蒽酮 J (82%0 = 164公斤联苯绕蒽酮 J (100%)
收率: 82.3%.
操作要点及不正常原因:
从操作难点到投料要点, 共14点: 略!
生产控制; 略! 原料控制: 略! 成品控制: 略!.
5.
联苯绕蒽酮 J [116-90-5].
生产工艺. FIAT 1313. II. 81-5. 英文, 摘译自PB. 报告. 略!.
二羟基紫蒽酮. [128-59-6] 生产工艺. 1931年5月28日. 译自PB. 25624. 1021 -
30
在3.2立方米铸铁锅内, 加入400公斤96%硫酸, 于室温加入150公斤联苯绕蒽酮, 100%计, 此时自行
升温到30 – 35℃,
在冷冻盐水冷却下,
迅速加入500公斤水, 温度不宜超过30 – 40℃
(取样
1), 加完冷却到15
– 20℃, 在同时冷却下, 于2小时内加入220公斤天然二氧化锰, 温度勿超过30℃,
加完二氧化锰,
在20
– 25℃, 保温反应4小时以上 (取样 2), 反应完毕, 用二台8平方米的吸滤器过滤, 压紧, 抽干, 用1300公斤硫酸 (600波美) 分两次洗涤 (取样 3), 滤饼在20立方米的衬砖锅内, 加15立方米热水和950公斤左右的亚硫酸氢钠溶液, 用直接蒸汽加热煮沸2小时, 到不再有亚硫酸氢钠为止 (取样 4), 用木压滤机 (1400 x 1400, 36框) 压滤, 用热水洗到无酸性 (取样 5), 滤饼于130℃减压干燥 (取样 6和7).
得量: 约165公斤二羟基紫蒽酮 (95%) = 157公斤 (100%).
收率: 98%.
生产要点:
加二氧化锰, 温度不能超过30℃.
氧化滤饼一定要用600波美硫酸彻底洗涤, 以除去杂质.
要将过氧化的醌体还原, 必须使用足够量的水和亚硫酸氢钠煮沸处理.
过滤以及洗涤时的滤饼, 勿用压缩空气吹, 以免产生醌体, 滤饼干燥时, 必须避免与空气接触.
不正常原因:
酸度太高. 反应时间不够 (加完二氧化锰, 至少反应4小时), 会使收率降低.
滤饼洗涤不彻底, 会使质量下降, 洗涤后的滤饼必须用足够量的水和亚硫酸氢钠煮沸处理, 否则锰不能以硫酸锰的形式除去.
滤饼用空气吹或干燥时接触空气, 都会产生醌体紫蒽酮.
生产控制分析:
取样 1. 联苯绕蒽酮溶解终点. 加水到630波美 (84%硫酸)后, 取样于瓷皿内或者在显微镜下观察, 应完全溶解, 个别絮状沉淀, 可勿略不计.
取样 2. 氧化终点. 反应四小时后, 由氧化物料取样.
2.1: 于显微镜下观察, 应为长形斜断面深色结晶, 其间夹有无色硫酸锰结晶 (为六角形片状) 和深色小块 (未反应的二氧化锰), 母液应不再有红色和不透光性, 而呈蓝紫色, 透光.
2.2: 取小样 (1/10 g), 于30毫升稀亚硫酸氢钠液中, 煮沸数分钟, 过滤, 用热水洗涤, 滤渣于陶瓷器中干燥, 取样溶于浓硫酸, 于标准溶液比较, 必须为蓝紫色, 红色表明有未反应的联苯绕蒽酮.
如亚硫酸氢钠还原不够, 溶液呈棕红色, 可加少量结晶氢醌, 使醌体迅速还原.
取样 3. 酸化终点: 经过滤, 洗涤, 最后的洗涤滤液应呈红棕色, 加水有少量沉淀析出, 吸滤应无残留结晶. (青铜色).
取样 4. 亚硫酸氢钠煮沸终点 (还原终点): 测定应不再有亚硫酸气味 (于浓硫酸中显色, 看是否还有醌体).
取样 5. 滤饼洗涤终点:
5.1: 显微镜下应为均一的长形深蓝绿色斜断面结晶, 具有青铜光泽.
5.2: 滤饼于瓷干燥器中干燥, 测定锰含量.
5.2.1: 用碳酸钠 – 硝酸钾熔融, 应仅显微绿色.
5.2.2: 取0.2克二羟基紫蒽酮, 用1毫升浓硫酸 (98%)溶解, 于水浴上加热2 – 3分钟, 再用10毫升水稀释, 煮沸, 过滤. 将浅黄色滤液, 用少量浓苛性钠中和成碱性, 然后用0.5毫升双氧水 (3%)处理, 当有锰存在时, 析出棕色沉淀, 沉淀量应很少 (少数絮状物).
取样 6: 取减压干燥后的滤饼10克, 按一般方法甲基化, 然后与还原艳绿FFB标准打样比较.
收量: 应为 9 – 9.2克.
取样 7: 纯度测定: 测干品灰分含量, 再由100%纯品甲基化收量之差, 计算纯度.
原料, 联苯绕蒽酮: (纯度约83 – 85%). 熔点: 314 – 316℃.
杂质: 苯绕蒽酮 约 3%. 灰份. 约4%. 其它. 约8 – 10%
96%硫酸中. 红色, 微带荧光.
成品, 二羟基紫蒽酮: (纯度约 95%). 其中灰份约 3 – 4%
96%硫酸中. 红紫色 染纸: 绿色遇酸变蓝.
生产周期: 24小时, 溶剂回收: 620波美废硫酸。 引用专利:DE. 411413; US.
1564423.
生产操作经验:
因为联苯绕蒽酮的氧化是在低温, 而且最终的硫酸浓度是84%, 所以氧化可以使用铁锅, 如果在96%硫酸中进行氧化, 那么, 反应可以进行得很快, 但在过量的二氧化锰存在下, 容易发生过氧化, 从而使收率降低, 产品质量低劣. 氧化前酸度控制到84%, 还能使一些已有的二羟基紫蒽酮立即以醌的型式析出, 从而减少二氧化锰的用量. 同时使灰份以外的所有杂质仍在溶液之中, 二氧化锰过量50%是必要的, 量少反应会不完全.
反应条件与反应范围: 在84%的硫酸中, 于15 – 20℃氧化4 – 6小时到反应终了. 当温度超过30℃,并且酸浓度大于86%时, 对反应不利. 反应温度低于15℃,
结晶出来的粒子细,
会很难过滤.
二羟基紫蒽酮. [128-59-6] 生产工艺. 1942年1月22日. 译自PB. 70057. 8452 –
4.
由于战争的关系, 二氧化锰缺货, 同时可供使用的二氧化锰纯度也只有60 – 70%左右, 这样一来, 一方
面要分离产品中的矿渣, 另一方面, 也要回收二氧化锰, 因此, 生产基本上是不定期的.
生产设备: 略!
生产工艺; (操作步骤).
在3.2立方米铁反应锅内, 先加入2000公斤96%硫酸和2500公斤75%硫酸, 配成4500公斤84%硫酸, 而后加入155公斤联苯绕蒽酮 (100%计), 搅拌4 – 5小时使其溶解, 再将其冷却到15℃, (在2 – 4小时内)
通过5
毫米孔径的篩网加入192公斤二氧化锰 (100%计), 其中2/3 在15 – 25℃加入, 1/3 在25 -35℃加入, 加完, 在35℃保温2小时, 压料到铺有Vinoflex (氯乙烯与异丁氧基乙烯共聚物) 滤布的衬胶压滤机内压滤.
滤饼 (二羟基紫蒽酮 + 硫酸锰 + 矿渣), 于3.2立方米铁酸处理锅内, 用由1722公斤96%硫酸和1278公斤24%发烟硫酸配成的3000公斤100%硫酸, 于室温搅拌到二羟基紫蒽酮溶解为止 (4 – 6小时), 压料到每台7平方米过滤面积的2台衬砖吸滤器内吸滤, 再用3000公斤左右的96%硫酸洗涤 (滤渣处理见后).
由吸滤器出来的滤液, 压到10立方米的衬铅稀释锅内, 用水稀释到500波美的酸度, 冷却到40℃,
于铺有Vinoflex滤布的衬胶压滤机内压滤.
滤饼于配料盘内搅和, 集中三批, 于24立方米衬砖锅内, 加入15立方米水和1200公斤380波美的亚硫酸氢钠溶液, 用直接蒸汽加热煮沸4小时, 到检定不再有亚硫酸氢钠和醌体存在为止.
还原完毕, 用大型压滤机压滤, 滤饼洗涤到中性, 为了避免醌体的生成, 不能用压缩空气吹.
成品送611车间, 在130℃减压干燥.
得量: 161公斤.
(100%计). 收率: 98%.
二氧化锰的回收:
滤渣 (硫酸锰 + 矿渣, 约2500公斤), 于24立方米设备内, 加15立方米水溶解, 于铺有Vinoflex滤布的大型压滤机内过滤, 不溶物为矿渣, 滤液于24立方米衬胶锅内, 用4000 – 5000公斤400波美苛性钠中和到中性, 加入560公斤次氯酸钠, 搅拌一小时, 过量氯用亚硫酸氢钠破除, 回收得到的二氧化锰, 用压滤机压滤, 洗涤到硝酸银反应消失为止, 用箱式烘箱烘干.
回收量: 162 公斤. 回收率: 84.5%
8.
二羟基紫蒽酮. [128-59-6]
生产工艺. FIAT. 1313. II. 85 – 6. 英文, 摘译自PB. 报告. 略!
上海染料生产工艺汇编.
395 – 7 (1976). 其中[116-90-5]已在2018年4月17日上网。
抄录说明:
在现代化,科技进步的今天,由于有那么多的新技术,又是什么“黑科技”等。好像有点多余。但是,我想总比当废纸处理好多吧!所以仍在每天上网抄录一点资料。请读者理解,理解万岁!
日本人的译文。
(日)细田豊 《理论制造染料化学》。技報當 出版。 1957年。 P. 721-722. CAS号[116-90-5]和[128-59-6],抄录如下:
(1)2,2’-Dibenzanthronyl.[116-90-5]。90% KOH 500 kg,NaAc 60 kg,イソブタノ-リ385 kgを140-1450に加热して溶解し,1000に冷して升华ベンサントロン200 kgを10 hかかつて加え最後に1120となるようにする。1120で搅拌,イソブタノ-ル100 kg追加,900に冷して水1 tを加え25-300まで冷却し,静置して下层のKOH溶液1 m3を引拨き,残る上层のbenanthronyl およびそのリウコ体とイソブタノ-ル混合物に水400 lを加え,水600 lとNaOCl 12% 液80 kgの混液を1 hで加え1/3 h搅拌後加热してイソブタノ-ルと水の共沸混合物を蒸馏し分离水は釜に戾しながら3 hで33%のイソブタノ-ルルが回收される。つぎに水8 tに出し生蒸汽で90-950に上げ滤過,湯洗,亁燥する。200 kg (82%) = 164
kg (100%), 收率 82.3%。
(2)Bz-2-Bz-2’-Dihydroxydibenzanthrone.
[128-59-6]。 84% 硫酸4.5 tにジベンザンスロニル190 kg (82%)を加え300で4-5 h搅拌後150にてMnO2 192 kg (100%) を5 mmの筛を通して2/3 は15-200で1/3 は25-350で加え,25-300で2 h 搅拌,滤過し,ケ-クを99.5% 硫酸3 tに溶解し7 m2 stone filter 2台で滤過,96% 硫酸3 tで洗う。滤液を500Be’ までうすめ400に滤過し,3回分のケ-クに15 m3の水およびNaHSO3 380Be’ 1.2 tを加え4 h 煮沸し,滤液し减压亁燥する。161 kg (100%), 收率94%。
抄注:未注明译自哪个PB报告。
(俄)A.B. Ельцова。《染料及中间体实验室合成方法》。1985年。§2.6 还原亮绿C。 16,17-二羟基紫蒽酮。[128-59-6].
预先准备二氧化锰悬浮液:在100毫升烧杯中加入8.6克二氧化锰,然后加入20毫升浓硫酸,用玻璃棒搅拌成均匀的悬浮液。
将装有搅拌和温度计的500毫升三口烧瓶,置于电加热水浴中,加入115毫升92% 硫酸,搅拌下加入102克联般若堂,加料速度控制物料温度不超过35℃,搅拌到完全溶解(约2小时),冷至15-20℃,慢慢加入上述制备好的二氧化锰悬浮液,加完的温度应不超过34℃,保温反应30分钟,用硫化钠测氧化反应终点:取几滴反应物料,置于加有6毫升76%硫酸的试管中,搅拌後分装成2个试管,一个加入少量无水硫化钠,摇匀后将2个试管进行比色,加有硫化钠试管的液体应为无色,如仍为红色,反应物中再加入0.5克固体二氧化锰,于34℃再保温30分钟,然后在34-36℃加入1.3克甲醛,此时未反应的活性二氧化锰被甲醛还原,它还可预防二羟基紫蒽酮过氧化。反应物料用布氏漏斗过滤,深棕色滤饼置于150毫升烧杯中,加入55毫升浓硫酸,用玻璃棒搅拌使其溶于酸中,必要时可将酸液进行过滤,滤液置于加有1.5毫升水的150毫升烧杯中,稀释后(酸含量约为77%),物料成沉淀析出,再过滤,滤饼用水(每次25毫升,总量100毫升)洗涤,抽干,得深灰色16,17-二羟基紫蒽酮氯苯,其中可能含有5,10,16,17-四氧-5,10,16,17-四氢紫蒽酮。
纯度测定:取少量成品溶于乙醇,从中取出少数溶于滴于层析硅胶板上,用甲苯:乙醇 = 7:4展开剂展开,二羟基紫蒽酮,Rf 0.65; 二氧紫蒽酮,Rf 0.22.如果成品中混有二氧紫蒽酮,需用亚硫酸氢钠处理。
将装有搅拌,温度计和回流冷凝器的200毫升三口烧瓶,置于电加热油浴中,加入混有少量二氧紫蒽酮杂质的二羟基紫蒽酮滤饼,搅拌下慢慢加入15毫升 38% 亚硫酸氢钠溶液,加热到80℃,搅拌2小时,根据二氧化硫过量情况定还原终点:取反应物料滴于玻璃板上,相聚1厘米处放置,用0.1 M碘溶液润湿过的刚果红试纸,料液接触后试纸上无深蓝色出现,证明还原完毕,物料移至300毫升烧杯中,加入200毫升水,用玻璃棒搅拌均匀,过量,氯苯永约500毫升水(每次50毫升)洗到中性,抽干,置于培养皿中,于100℃烘箱中干燥。
得量:9.4克(82%)。产品为黄灰色粉末。 (抄注:译文仅供参考!)。
国内出版物。
赵维绳 陈 彬 汪维凤 编著。染料丛书《还原染料》。化学工业出版社 出版。1993年。
抄注:陈 彬 老先生是早期留德的染料专家。1947年编写有《还原染料》一书。回国后在沈阳院工作。
对于本产品,第261写有16,17-二羟基蒽醌,但无生产工艺文献说明。第266页(1)制联苯绕蒽酮。完全抄自[J] 有机化学工业技术报导, 1958, 10, 31。但是未说明资料来源。
全书引用的历史文献只有BIOS和FIAT,就是没有早期进口的PB报告!
何岩彬 主编 《染料品种大全》。沈阳出版社 出版。 2018年。 “染料中间体及可合成的染料”p. 1851-2038.
本产品请见p. 2038.
陈忠源. 2019年9月14日星期六。






![C.I. 酸性蓝145 (C.I. 62070) 生产工艺 CAS号[6408-80-6]](/chenzhongyuan/Upload/ArticlePicture/IMG_29062016_105104.png "C.I. 酸性蓝145 (C.I. 62070) 生产工艺 CAS号[6408-80-6] 2016/6/29")
![C.I. 酸性橙51 (C.I. 26500) 生产工艺 CAS号 [8003-88-1]](/chenzhongyuan/Upload/ArticlePicture/IMG_21062016_132801.png "C.I. 酸性橙51 (C.I. 26500) 生产工艺 CAS号 [8003-88-1] 2016/6/21")
![CAS号 [109-89-7] 生产工艺。 二乙胺](/chenzhongyuan/Style/Blue/Style_Bullets_Picture.gif "CAS号 [109-89-7] 生产工艺。 二乙胺 2020/7/20")
![CAS号[74920-95-9] - C.I.酸性黑71](/chenzhongyuan/Upload/ArticlePicture/QQ图片20210721154451-1.png "CAS号[74920-95-9] - C.I.酸性黑71 2021/7/21")
![CAS号 [130-00-7] 生产工艺。 1,8-萘内酰胺](/chenzhongyuan/Upload/ArticlePicture/QQ图片20201228101820.png "CAS号 [130-00-7] 生产工艺。 1,8-萘内酰胺 2020/12/28")