CAS号 [89-41-8] 生产工艺 3-硝基-4-甲氧基苯甲酸
CAS名: Benzoic acid, 4-Methoxy-3-nitro-, 参考文献: Beil. 10, 181.
用途:有机合成和红色有机颜料,请见红色基KD和红色基KL等生产工艺。
生产工艺文献: 本人根据特种文献整理如下。附加国内研究开发动态。
BIOS 1153, 46-48. (=胶卷PB 85687) 3-Nitro-p-anisic acid (Leverkusen) 英国人译自德文。抄录如下。
反应式: 本人有加注。译者未说译自哪个PB报告?这是路线1.
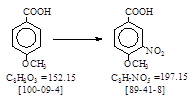
Plant: 1 C.I. nitrator, 3000 l., with
lid and stirrer, cooling jacket coils for brine cooling.
1 8000 l. tiled vat with lead coils and brine connection. 1
Tiled nutsche.
1 Tiled blow-egg. The tiled units are jointed with asplit. 2
Wooden vats, 8000 l. 1 Small rubber or lead press.
Materials: p-Anisic acid
M.W. 152 200 kg. (100 = 45.4 kg. NaNO2) Sulphuric acid 98% 2000 kg.
Water 40 kg. Mixed acid N (33.0% HNO3 , 48.0% H2SO4 , 19% H2O ) 260 kg.
Process: (1) Nitration:
The nitratot is charged with 2000 kg. of 98% sulphuric acid and 40 kg. of water, followed by 200
kg. of p-anisic acid (dried and ground) at 5-100C.
As soon as solution is complete, mixed acid N (260 kg. =97% theory) is run in with very good
agitation below 00C. It
is then stirred for 12 hours. Test:
100 g. of the nitration are dissolved in 1000 cc. water, reduced with zinc
dust and estimated with nitrite. The requirements should be not less than 35.4 g NaNO2 (theory 36.2). The m.P. of the precipitated nitrocompound should be 184-1850C.
The batch is now blown into 3000 l. of water, cooled with brine, so that the temperature does not
rise above 250C. Test:
1 l. filtrate from filtered product is reduced with zinc dust. The whole filtrate should be
equivalent to ca. 3 kg. NaNO2.
It is now dropped to a nutsche and washed with
water. (2) Purification:
The crude nitroanisic acid is stirred into 1000 l. of water at 500C and
nearly neutralized with 400 kg. of
caustic liquor followed by soda. It should give a clear solution. 10 kg of sawdust and 5 kg soda of desolourising carbon are added and the batch screened
through a small press into a second vat. Here it is acidified to Congo paper
with 300 l. of hydrochloric acid and
the pure white product filtered at 250C, washed a little and finally hydro-extracted. Yield = 432 kg moist. It is dried at 70-750 and ground.
Yield =
225 kg (100= 33.0 g NaNO2 by NO2and 100= 33.2 g NaNO2 by COOH.). = 74 kg NaNO2 = 74 kg NaNO2 =
82% theory.
M. Pt. 184-1860 (Lit. 186-1870).
Notes: A
yellowing of the product indicates some hydrolysis
of the methoxy-group. To prevent this hydrolysis the sulphuric acid must
not be higher than 96%. It is also
desirable to use a mixed acid containing 20%
water.
To prevent sulphonation the addition of p-anisic acid must be made below 100 and the nitration
started immediately solution is complete.
FIAT 1313,I, 206-7. (=胶卷PB 85172) 101. 3-Nitro-p-anisic acid. (I.G.
Leverkusen) o-nitroparanisaeure. 美国人译自德文,抄录如下。
译者也未说译自哪个PB报告?基本上是同一德文生产工艺,只是译法不同,但本人现在未找到原件。
Apparatus: 1 – cast iron
jacketed nitrator, with internal cooling coil, 3000 l. 1 – stoneware nutsch.
1 – stoneware receiver. The stoneware
apparatus jiones are sealed with cement.
2 – wood tubs, 8000 l.
1 – small press, lead or rubber-lined.
Materials: 1 – 200 kg
anisic acid (45.2 NaNO2/100) 2 – 2000 kg H2SO4 98% 3 – 40 kg
H2O.
4 – 260 kg mixed acid (33 HNO3 , 48 H2SO4 , 19 H2O )
Charge nitrator with (2) and (3) and cool to 5 to 100C. Add (1) and let
dissolve. Cool to under 00C. and
add (4) gradually, keeping temperature at 00C. or under. Stir 12 hrs. Test by
reducing a drowned sample with zinc
dust. 100 parts should consume 35.4 parts NaNO2. Theory = 36.2
parts NaNO2 . Product melts at 184-50C. Drown nitration in 3000 l. brine-cooled
water, keeping temperature no higher than 250C. Test: 1 l. of
suspension reduced with zinc consumes about 3 kg NaNO2 . Filter on nutsch and wash.
Purification:
Dissolve the crude in 1000 l. water at 500C. with about 400 kg. NaOH and then Na2CO3 . Obtain nearly neutral clear solution. Add about 10 kg. saw-dust and 5 kg. decolorizing carbon. Filter through small
press to 2nd tub. Add about 300
l. 200Be’ HCl to make acid to Congo. Filter on nutsch at about
250C. and wash. Centrifuge cake from nutsch. Yield: 432 kg. wet product.
Dry at 70-750C. and mill. Yield = 225 kg. dry
product, analyzing 33.0 NaNO2 /100 for nitro group and 33.2 NaNO2/100 for COOH group. = 82% of theoretical. Melting point = 184-60C.
Lit. 186-70C.
Notes
on process: Yellow coloration of product is caused by hydrolysis
of the OCH3 group. To prevent this the H2SO4 must be at least 96%. It is advantageous to use mixed
acid with 20% water content. To prevent
sulfonation do not dissolve (1) about 100C. Start nitration at once after (1) is dissolved.
中文摘译。张澍声。《精细化工中间体工业生产技术》1996年。P. 95.(译自BIOS和FIAT)请见原书。
国内研究动态:
吕玉芝 唐会林 红色基KD合成新工艺探讨[J]染料工业,1996,2, 37-38(32) 2. 硝化 抄录如下。
在500 ml的三口瓶中加入4-甲氧基苯甲酸41.5 g , 二氯乙烷166.0 g , 98%硫酸o.5 g。 在450C下,于3小时内滴加98%硝酸19 g . 在45±20C, 保温2小时。经后处理,得产品3-硝基-4-甲氧基苯甲酸52.7 g , 重量收率98%。上文未引用上述文献。
反应式: 本人有加注。 这是路线2。

FIAT 1313,I, 216-7. (=胶卷PB 85172) 109.
3-Nitro-4-methoxybenzyl chloride. I.G. Hoechst
1947年6月10日。美国人抄自德文。 “Nitroxyl chloride” Apparatur: 250
lose verbleiter Kessel, Ruehrer 100 Touren, Tonnutsche.
Einsatz: 82 kg Schwefelsaeure 95%ig. 50 kg Formaldehyde 30%ig. 50 kg
Nitroanisol. 32 kg
Salzsaeuregas.
Im Kessel, der durch eine hinter den Kuehler
geschaltete Waschflasche mit Schwefejsaeure abgesperrt ist, werden zu vorgrlrgten 50
kg Formaldehyd 30%ig unter Ruehren und Kuehlung bei 15-200 innerhalb 4-6
Stunden mittels Tropftricher 82 kg
Swefeksaeure 95%ig zugegeben. Dann leitet man bei 20-250 Salzsaeyregas in einer stuendlichen Menge von etwa 1 kg bis zur Saettigung ein (Dauer etwa 10 Stunden). Anschliessend laesst man innerhalb 1 Stunde bei 200 50 kg
Nitroanisol zulaufen und setzt dann die Einleitung von gasfoermiger Salzsaeure – stuendlich 1/2 – 1 kg – bei 500 nochmals etwa 10 Std. fort. Das ende der Umsetzung wird an folgenden Proben feztgestellt (抄注: 打字错了,应是 festgestellt !)这里说明资料是按德文原件打字的!
Eine im Labor abgesaugt Probe darf in der Lauge kein Oel
mehr enthalten und der Schmelzpunkt der
rohen Ware soll bei 85-860 leigen. Wenn diese Bedingungen noch nicht erreicht sind, muss weiten
Salzsaeuregas eingelegtet werden. Der Kesselinhalt wird nach Beendigung der
Reaktion abgekuehlt, abgesaugt, der Rueckstand neutral gewaschen und im Vakuum bei 40-500 getrocknet.
Ausbeute: 65.3 kg
Qualitaetspruefung: 10 g werden
mit 10 ccm Sprit und 6 ccm Pyridin eine halbe Stunde auf dem Wasserbad erwaermt, dann mit
Wasser verduennt und abgesaugt. Der gewaschnene und
getrocknete Rueckstand (vorwiegend Dinitro-dimethoxy-diphenylmethan)
Soll nicht
ueber 5% betrageb. Der Schmelzpunkt der
reinen Substanz ist 87.50 . 说明反应物中有3,3‘-二硝基-4,4‘-二氨基二苯甲烷。
这是3-硝基-4-甲氧基氯苄 [6378-19-4]的生产工艺。
中文摘译。张澍声。《精细化工中间体工业生产技术》1996年 P. 154. 请见原书。
国内开发申请的专利:
张正富 等。中国专利 CN 104356000. 2015年2月18日。未提有上述文献。部分抄录如下。
(1)制备3-硝基-4-甲氧基氯苄。在500 ml四口反应瓶中开动搅拌,将154.9克98%硫酸溶液加入40.6克,30%的甲醛溶液中,再加入75.5克,30%盐酸,再加入折百68.4克 (0.4 mol)邻硝基苯甲醚,在65-700C, 压力0.1 Mpa下,保温反应10小时,反应完成后,将反应液冷却至150C, 过滤得到3-硝基-4-甲氧基氯苄产品79.9克,含量99.3%。收率90.3%。=[6378-19-4]
(2)制备3-硝基-4-甲氧基苯甲基醇。 在500 ml四口反应瓶中,将步骤(1)所得3-硝基-4-甲氧基氯苄固体加入到400 ml, 15%氢氧化钠水溶液中,开动搅拌升温至800C, 中和反应约12小时,反应结束,过滤得3-硝基-4-甲氧基苯甲基醇产品69.6克,含量99.1%,收率95.1%。 =[41870-24-0]
(3)制备3-硝基-4-甲氧基苯甲酸。 在四口反应瓶中将所得3-硝基-4-甲氧基苯甲基醇加入到300 ml, 60%浓度的硝酸溶液中,开动搅拌慢慢升温进行反应,在温度85-900C, 压力0.1 Mpa下,保温反应10小时,反应结束,过滤得产品3-硝基-4-甲氧基苯甲酸产品67.9 g,含量99.3%,收率91.2%。为白色晶体,熔点≥1910C. 收集滤液备下一批套用。=[89-41-8]
加注:
1. 同一德文生产工艺,英美译法不同,最好是看原文。问题是我还未找到原文,所以没有抄录。
2. 在2015年的中国专利中,未见到对产品的历史进行评述,我认为1996年,染料工业,1996,2,37-8.
吕玉芝和唐会林同志的评述是正确的,问题是二者均未引用早期德文生产工艺这一历史事实。又因为国内外没有此类文献的编目和收录。我想,专利局是会容易通过的,是否实用请读者评述!
3. 由于本人未收录其它PB报告目录,所以,3-硝基-4-甲氧基苄醇的德国生产工艺原文未抄录。
4. 我已发现美国人抄录的德文,因打字原因,有打错的。前面也提到过。
5. 本人再次提出,有一些历史文献,对我们的开发还是有用的,希望能利用。
陈忠源 2016年11月27日 于 无锡 明辉国际,






![C.I. 酸性蓝145 (C.I. 62070) 生产工艺 CAS号[6408-80-6]](/chenzhongyuan/Upload/ArticlePicture/IMG_29062016_105104.png "C.I. 酸性蓝145 (C.I. 62070) 生产工艺 CAS号[6408-80-6] 2016/6/29")
![C.I. 酸性橙51 (C.I. 26500) 生产工艺 CAS号 [8003-88-1]](/chenzhongyuan/Upload/ArticlePicture/IMG_21062016_132801.png "C.I. 酸性橙51 (C.I. 26500) 生产工艺 CAS号 [8003-88-1] 2016/6/21")
![CAS号 [109-89-7] 生产工艺。 二乙胺](/chenzhongyuan/Style/Blue/Style_Bullets_Picture.gif "CAS号 [109-89-7] 生产工艺。 二乙胺 2020/7/20")
![CAS号[74920-95-9] - C.I.酸性黑71](/chenzhongyuan/Upload/ArticlePicture/QQ图片20210721154451-1.png "CAS号[74920-95-9] - C.I.酸性黑71 2021/7/21")
![CAS号 [130-00-7] 生产工艺。 1,8-萘内酰胺](/chenzhongyuan/Upload/ArticlePicture/QQ图片20201228101820.png "CAS号 [130-00-7] 生产工艺。 1,8-萘内酰胺 2020/12/28")